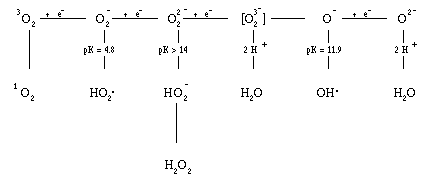
Figure 1. The partial reduction products of oxygen.8
_____________________________________________________________
O2- + O2- + 2 H+ ---> H2O2 + O2 (1)
The spontaneous rate for this reaction at physiological pH is 5 x 105 M-1s-1, and the enzyme superoxide dismutase, which catalyzes this reaction, increases it by a factor of 104.44 Thus, much of the damage that has been attributed to O2- may actually have been caused by H2O2 that was formed from the O2- that was initially generated.
O2- + H2O2 ---> H2O + OH- + OH. (2)
This reaction has become known as the Haber-Weiss reaction. Hydroxyl radical, too, could be the source of part of the damage that has been attributed to O2-. However, the Haber-Weiss reaction as written is exceedingly slow, with a reaction rate of 3.0 M-1s-1 74 at pH 7.3. Chelated iron, on the other hand, which is frequently present in biological fluids, catalyzes this reaction, increasing the reaction rate to a significant degree8,75. Iron is chelated in vivo with phosphate esters such as ADP, ATP, GTP, and pyrophosphate76, and other compounds such as citrate and oxalate77. In vitro chelators such as EDTA and nitrilotriacetic acid (NTA) also allow iron to catalyze the reaction. In contrast, chelators that prevent iron catalysis of reaction (2) are diethylenetriaminepentaacetic acid (DTPA),78 bathophenanthroline sulphonate (BPS),79 phenanthroline,79 and bipyridine.80 The reaction for the iron catalyzed Haber-Weiss reaction is as shown in equations (3) and (4).44
Fe+++ + O2- --> Fe++ + O2 (3)
Fe++ + H2O2 --> Fe+++ + OH- + OH. (4)
The rate constant for equation (3) when the iron is chelated with EDTA has been determined to be k2 = 1.3 x 106 M-1s-1 at pH 7.0.35 The rate constant for equation (4), which is known as the Fenton reaction, for EDTA-chelated iron is given as k3 = 104 M-1s-1 at pH 7.081. Thus, the catalytic properties of iron make the Haber-Weiss reaction quite possible in vivo and the hydroxyl free radical (OH.) is probably formed in vitro and in vivo mainly through the iron-catalyzed Haber-Weiss reaction (reactions 3 and 4).
Table I. Bimolecular Rate Constants for Reactions Between Scavengers
and the Hydroxyl Radical and Singlet Oxygen.
_____________________________________________________________
Scavenger Rate Constant (M-1sec-1)
_____________________________________________________________
Hydroxyl Radical92 Singlet Oxygen100,101
_____________________________________________________________
Sodium Azide 7.5 x 109 5 x 108
Dimethylsulfoxide 7.0 x 109 ~ 103
Sodium Benzoate 6.0 x 109 3 x 103
Thiourea 5.0 x 109 8 x 105
Sodium Formate 2.8 x 109 ~103
Ethanol 1.5 x 109 ~103
Urea ~106 ~103
_____________________________________________________________
(DMSO). The latter two scavengers are particularly useful for in vivo experiments, as they are capable of crossing cell membranes. Table I gives a more complete list of OH. scavengers, along with the rate constants of their reaction with OH.. Urea, a poor OH. scavenger, is frequently added as a control; the rate of its reaction with OH. is less than 7.0 X 105 M-1s-1. Some OH. scavengers, such as azide, also scavenge 1O2, but DMSO and ethanol only react with OH..
Defenses against Oxygen toxicity
In vivo methods of studying the photodynamic effect can make great use of
manipulations of the cell's natural defenses. Discovering the necessary natural
defenses also yields information on what forms of damage and damaging agents
contribute to lethality.
Cells maintain a variety of defenses against oxygen toxicity. Among these are an array of enzymes that have evolved to deal with oxidative stress, including superoxide dismutase and catalase (described below).44 In animals, additional defensive enzymes include methionine sulfoxide reductase, which repairs methionine residues in proteins that have been damaged by OH., glutathione peroxidase, and the glutathione reductase which regenerates the cofactor for the glutathione peroxidase. Still other defensive enzymes are endonucleases, exonucleases, and DNA polymerase, which repair single-stranded breaks and modified bases in DNA caused by oxidative and other stresses. In eucaryotes, uric acid, a-tocopherol, ascorbic acid, and b-carotene are among the compounds that function in vivo to scavenge active oxygen species and other organic radicals formed in the cell by reactions with the oxygen radicals; both procaryotes and eucaryotes contain high levels of glutathione, a scavenger of OH. and 1O2.44
O2- + O2- + 2 H+ ---> H2O2 + O2 (1)
Superoxide dismutases are ubiquitous in aerobic organisms.102 Eucaryotes contain, typically, both a copper and zinc-containing superoxide dismutase (CuZnSOD) and another SOD that contains manganese(MnSOD); the two are unrelated, and the latter is compartmentalized in the mitochondria, while the former is in the cytoplasm103. Bovine copper-zinc SOD has a molecular weight of 32,500104 and contains two atoms of copper and two atoms of zinc per molecule.105 It is composed of two identical subunits joined by at least one disulfide bond.105 The human CuZnSOD is similar to the bovine CuZnSOD.106 The mechanism of action of CuZnSOD involves alternate reduction and reoxidation of the Cu2+ at the active site during successive interactions with O2-.102 The mitochondrial SOD, MnSOD, contains four subunits, each with a molecular weight of 20,000; it is located in the matrix of the chicken liver mitochondrion.102
Procaryotes such as E. coli contain both an MnSOD and an iron-containing superoxide dismutase (FeSOD). The procaryotic MnSOD shows homology to the eucaryotic enzyme but contains two subunits rather than four and is consequently half as large, with a molecular weight of 40,000. The FeSOD is partially homologous to the MnSOD.107 It has two subunits, one Fe3+ ion per molecule of enzyme, and a molecular weight of 39,000. The mechanism of the Mn and FeSODs is probably similar to that of the eucaryotic CuZnSOD.102 Superoxide dismutases increase the spontaneous rate of O2- removal according to equation (1) from about 5 x 105 M-1s-1 at physiological pH44 to about 1.8 X 109 M-1s-1 for the MnSOD at pH 7.8 and about 1.6 x 109 M-1s-1 for the CuZnSOD between pH 5.3 and 9.5.44
Compounds that are capable of entering cells and inducing superoxide dismutase include paraquat (methyl viologen), pyocyanine, phenazine methosulfate, streptonigrin, juglone, menadione, methylene blue, and azure c, all known redox-cycling compounds.109
Low levels of the MnSOD are characteristically seen when cells have been grown in a glucose minimal medium. High levels of SOD are produced by growth in TSY, or by growth in minimal medium to which 100 mm iron(II), 100 mm manganese(II), 100 mm 8-hydroxyquinoline, and 10 mm paraquat have been added; levels are enhanced thirty-fold by the latter treatment.108 Similar induction levels are observed by the addition of paraquat and manganese alone at similar levels.
2 H2O2 ---> 2 H2O + O2, (5)
whereas peroxidase catalyzes the reaction:
AH2 + H2O2 ---> A + 2 H2O (6)
where A is an electron donor such as dianisidine or guiacol.110
There are two main enzymes with catalase activity in E. coli; one, hydroperoxidase II (HPII), possesses only catalatic activity. The other enzyme, which migrates more slowly than HPII during electrophoresis, hydroperoxidase I (HPI), is both a catalase and a broad spectrum peroxidase. When electrophoresed on a polyacrylamide gel, HPI can be visualized as two isozymes, HPI-A and HPI-B.111 Peroxidase (HPI) has a molecular weight of 337,000, is composed of four subunits of equal size, and contains two molecules of protoheme IX per tetramer.110 Catalase (HPII) has a molecular weight of 240,000 and consists of four polypeptides, each of which is associated with a ferric protoporphyrin IX.112
Low levels of catalase are observed in cells that have been grown in glucose minimal medium; levels four- to five-fold higher are seen when cells have been exposed for thirty minutes to 0.5 mm hydrogen peroxide or 5 mm ascorbic acid.111 The increase in catalase level is due to the induction of hydroperoxidase I, the broad spectrum peroxidase of E. coli. Enhancement of intracellular catalase is also seen when cells are pretreated with paraquat and manganese117.
Kinds of DNA damage that happens under the influence of light include altered or missing bases, single-strand breaks, double-strand breaks, and cross-linking. At least three kinds of damage are commonly defined: far ultraviolet kill, which seems mainly to result from the formation of thymine dimers; chemically-assisted ultraviolet kill (with psoralens for example), which results in DNA cross-linking; and the kind of kill seen as a result of exposure to near ultraviolet light or visible light in the presence of dyes, photodynamic kill, which includes modified bases, apurinic/apyrimidinic sites, and single stranded breaks. The differences in kinds of damage produced by these different forms of light were suggested by their being protected against by different repair systems.
Apurinic/apyrimidinic (AP) sites are generated spontaneously and by treatment with acid or as a result of chemical alkylation of deoxyguanosine, which weakens the N-glycosidic bond that attaches the base to the sugar-phosphate backbone of the DNA.123 They are also generated by DNA glycosylases, which recognize and cleave N-glycosidic bonds of damaged nucleotide residues in DNA.124
When the DNA backbone is broken by ionizing radiation or hydrogen peroxide, the ends contain 3' terminal phosphoglycolaldehydes125 or thymine glycols,126 or 3' terminal phosphates,125 which cannot act as substrates for DNA polymerase I or DNA ligase. These blocked 3' ends must be removed before repair of the DNA can take place, starting with the 3'-OH group of the primer DNA.127
DNA repair systems in E. coli include photoreactivation repair, excision repair, recombination repair, and SOS repair. Photoreactivation involves the cleavage of thymine dimers into a pair of normal thymines by photolyase, an enzyme activated by visible light.130
Excision repair is a process in which two cuts are made in the sugar-phosphate backbone of the DNA on either side of a distortion caused either by a thymine dimer or by a base with which no nucleoside can form a base-pair, producing a 3'-OH group on the 5' side. This allows DNA polymerase I to synthesize a new strand, displacing the defective strand; DNA ligase joins the new strand to the original strand. The original incisions are made in E. coli by endonuclease I. 130
Recombination repair, also called daughter-strand gap repair, involves by-passing a block to DNA polymerase III, allowing replication to continue, leaving a gap opposite the block (e.g., a thymine dimer). The unpaired gaps are filled by excising the homologous piece of undamaged sister strand and inserting it into the gap. DNA polymerase I and DNA ligase then join the inserted piece to adjacent regions and fill in the gap left in the donor segment.130
In SOS repair, binding of the recombination A (RecA) gene product to single-stranded DNA in the region of a thymine dimer or other distortion allows the insertion of adenines or mismatched bases by DNA polymerase III. The RecA protein also induces a series of other proteins, called din for damage inducible, which are kept repressed by the LexA protein in the absence of single-stranded DNA.130 A model of the SOS regulatory system is shown in figure 2. A fusion of one of the genes for a din protein and the coding region for the �-galactosidase gene has been made, allowing the induction of the SOS response to be easily studied by assaying for �-galactosidase levels after submitting the cells to an appropriate inducing stress agent, such as mitomycin c.131 It was thought at one point that the protein rec A protected against both far and near ultraviolet and photodynamic killing in E. coli;132 later studies showed that rec A did not protect at all against photodynamic killing, while nur showed great protection.115
Figure 2. Model of the SOS regulatory system. The open circles represent proteolytically inactive RecA molecules and closed circles represent proteolytically active RecA molecules. The semicircles represent LexA molecules. (From G. Walker, Microbiol. Rev. 48: 60-93.)
The gene for the E. coli HPI has been placed onto the plasmids pBT22 and pBT28 by Triggs-Raine and Loewen.119 The gene for the E. coli MnSOD was placed onto the plasmid pDT1-5 by Danielle Touati.122 Bernard Weiss's group placed the endonuclease IV (nfo) gene on a plasmid, pWB21.135 All three of these plasmids were used in this study.
The Photodynamic Effect
Oxygen-dependent toxicity that also requires the presence of light and a
sensitizing compound is known as the photodynamic effect.137 It was first described
by Raab in 1896.137 It affects all aerobic eucaryotic and procaryotic organisms and
has been the subject of thousands of studies. The mechanism of the photodynamic
effect remains mysterious138. Visible light is not absorbed by most cellular
components, and therefore does not usually have the toxic effects associated with
X-rays or ultraviolet light. However, a sensitizer that absorbs visible light-a colored
substance, or dye-may serve to transfer the energy of the light absorbed, usually
through oxidation or reduction reactions, to a species which will then cause damage
to cellular components. The wavelengths of light and their activities, as well as the
spectra of representative photosensitizing dyes, are shown in figure 3.
Photosensitizers are molecules which can absorb light to produce a chemical
reaction which would not occur in their absence.139
Figure 3. The phototoxic spectrum.
conditions that bring about the end of the toxic red tide, as photosensitizing compounds are released by the deaths of creatures killed by the red tide bloom.154
Figure 4. Structure of dyes.
Herrin (1982).181 The acridines studied include acridine orange (wavelength maximum 492 nm, extinction coefficient 41,600 M-1cm-1), acridine yellow (wavelength maximum 442 nm, extinction coefficient 29,000 M-1cm-1), and proflavin (wavelength maximum, 444; extinction coefficient, 34,300 M-1cm-1). Xanthenes included were fluorescein (wavelength maximum 496 nm) and rose bengal (wavelength maximum 550 nm, extinction coefficient 99,800 M-1cm-1), which possesses one of the highest absorption coefficients known.182 The phenazine studied was neutral red (wavelength maximum 540 nm). Thiazines included methylene blue (wavelength maximum 665 nm, extinction coefficient 78,000 M-1cm-1), azure c (wavelength maximum 616 nm), thionin (wavelength maximum 598 nm), and toluidine blue (wavelength maximum 626 nm).
Many, but not all, of these dyes are capable of penetrating into the procaryotic cell. Those dyes which cannot enter the cell will be able to damage only the cell membrane, which can itself cause cell death; however, in the absence of a reductant, dyes that cannot enter the cell are not lethal.183 The porin channels in the outer membrane of E. coli freely allow the passage of small positively charged molecules, but keep out large, hydrophobic or negatively charged compounds184 in order to exclude the hydrophobic and anionic bile salts that are a threat to E. coli in its natural habitat.185 In the drawing in figure 4, the thiazines and the phenazine have positive charges, while the other dyes are neutral. In experiments by Martin and Logsdon (1987), the dyes toluidine blue and acridine orange penetrated the cell, while fluorescein and lucifer yellow did not.183 Neutral red and methylene blue are accumulated in E. coli at neutral pH, but rose bengal only at acidic pH.186 In general, the
acridines, thiazines, and phenazine enter the cell, whereas the xanthenes and the napthalimide lucifer yellow do not.
Most of the dyes used in this study intercalate in between the bases of DNA. The acridines, thiazines, and phenazines all have planar, tricyclic structures, as seen in figure 3; however, the xanthenes are bulkier and cannot fit in between the bases of the DNA. In addition, the xanthines are negatively charged and are probably repelled by the sugar-phosphate backbone of the DNA. Xanthenes, unlike acridines, thiazines, and phenazines, do not intercalate into DNA. Since intercalating dyes will be largely intercalated into the DNA once they enter the cell, it is important to study dye reactions when the dyes are in the intercalated state, as well as alone.
photodynamic action. Although they vary widely in their structures they are all capable of oxidation reduction reactions, and, because of this fact, of giving rise to oxygen radicals.183 Most of them are known to generate singlet oxygen (1O2) as well.146 Extensive studies have been done illustrating the effects of these particular dyes in E. coli, before the significance of O2- was known. Many of these dyes are structurally related to known redox active compounds such as pyocyanine and phenazine methosulfate, which are known to give rise to O2-by redox-cycling within E. coli.187 Reasons why so many structural classes of dyes were considered in this study were in order to determine whether their individual toxicities could be explained by a common mechanism; to determine the relative importance of cell localization in determining their toxic effects; and to illustrate that all forms of light, from near ultraviolet through the red, are sufficiently energetic to promote phototoxic effects in the presence of an appropriate photosensitizing agent and to do so through the generation of reduced oxygen species.183
Proflavin plus light caused single stranded breaks in the bacteriophage FX174, where any single-stranded break is a lethal lesion. The packaging of DNA inside the phage head increases damage over isolated DNA.224 Strand breakage has been demonstrated to be caused by acridine orange,218 methylene blue,219 and ethidium bromide.225
Proflavin nicks inhibited template activity of DNA polymerase in FX174,223 indicating that the broken ends of the DNA do not contain a free 3'-OH group, which is required for the initiation of repair synthesis of the broken strand. The loss of infectivity of FX174 following photodynamic treatment with proflavin can be explained by a block in DNA polymerase reaction; termination occurs one base before a damaged guanine residue.226 The photodynamic reaction of methylene blue with deoxyribonucleic acid led to the rapid loss of ultraviolet absorbance accompanied by the uptake of one mole O2/mole derivative; the reaction occurred with the guanine compounds, while thymine compounds reacted very slowly227. Photodynamic reaction of methylene blue with DNA was more rapid with denatured than with native DNA.228 Strand scission of DNA in vivo has been shown for acridines with bacteria 229 and cultured mammalian cells.230 There is a peak in DNA damage for human cells but not B. subtilis when irradiated at 450 nm even without the addition of exogenous sensitizers, indicating that endogenous riboflavin probably acts as a sensitizer in the human cells.223
Mutation in E. coli to resistance to bacteriophage T5 was induced by visible light (>408 nm) and black light (300-400 nm), causing mutation rates to increase more than 18-fold. In Escherichia coli B strain S, mutagenesis was produced by acridine orange and proflavin, both of which are radiomimetic, i.e., they induce cross-resistance to ultraviolet radiation and to each other; this suggests a similarity in the mechanisms used to protect the cells against the two forms of damage. All radiomimetics are mutagens in E. coli B.236 Acridines induced mutation in E. coli tenfold greater than the normal rate in the absence of dyes.237
_____________________________________________________________
0dye + hv --> 3dye (7)
3dye + AH2 --> dye.- + AH. + H+ (8)
Type I
dye.- + O2 --> dye + O2- (9)
3dye + O2 --> dye + 1O2 (10)
Type II
3dye + O2 --> dye.+ + O2- (11)
1O2 + NADH --> O2- + NAD. (12)
_____________________________________________________________
Figure 5. Dye reactions. "AH2" represents a reductant; "3dye" represents a triplet state dye.
which secondarily damages the target; in either case, the initial reaction with the sensitizer is known as a Type I reaction (fig. 5). Alternatively, the triplet state photosensitizer may be quenched directly by oxygen, yielding either 1O2 or the O2- radical by electron transfer, in what is known as a Type II reaction (equations 10-11). For most sensitizer triplets, even in the absence of a reaction with oxidizable substrate, a small but significant percentage of interactions between the sensitizer and oxygen result in electron transfer, which produces O2-243. Either of the two dioxygen radicals, the singlet oxygen or the O2- anion, may then go on to react with a target molecule. Superoxide, as previously indicated, may dismute to form H2O2 and may ultimately give rise to the OH.. Both species exert deleterious effects in biological systems.
When Nilsson et al. (1978) observed that "until now there has been no conclusive evidence for the participation of singlet oxygen in any biological photooxidation in solution," they claimed that now they had unambiguous evidence for the participation of singlet oxygen in photodynamic oxidation of amino acids, but their unambiguous evidence turned out to rely on the observation of the destruction of histidine, since there is a low rate constant for reaction of histidine with triplet dye-without considering rate constants for alternative reactions of histidine with O2-, peroxide, or OH..263 Singlet oxygen has been assayed as the decrease in tryptophan absorbance at 280 nm, without confirming assays264, when clearly there are more species than singlet oxygen capable of degrading tryptophan, and some of them, such as OH., are very likely to be produced by singlet oxygen-generating systems. In fact, tryptophan degradation has been used as a direct assay for the production of OH.. Deuterium oxide enhancement is very small in toxicity in yeast with acridine orange, proflavin, and acriflavin, though somewhat larger for rose bengal.265 The role of singlet oxygen was investigated but could not be established in the methylene blue-photosensitized strand cleavage of DNA, because there was little increase in reaction in the presence of deuterium oxide.266
The formation of OH. by methylene blue in the presence of ascorbate has been observed.289 When exposed to the spin trap compound DMPO, photoexcited psoralens were found to produce a DMPO-OH EPR signal that is abolished when the reaction is carried out in the presence of OH. scavengers.168 Hydroxyl free radicals are produced by hematoporphyrin derivative, ascorbate, and light.290 In the dye-sensitized photooxidation in methanol of polyunsaturated fatty acids, using the dyes methylene blue, erythrosin, hematoporphyrin, and riboflavin, the isomer product distribution was interpreted in terms of a dual singlet oxygen and radical mechanism.142 In the photoactivated reaction of tartrazine, which is mutagenic only in the presence of light, there is no singlet oxygen participation, according to the cholesterol reaction assay, in which singlet oxygen is detected by its reaction with cholesterol, which results in reaction products specific to this reaction. However, superoxide dismutase & catalase inhibited OH. production, as measured by EPR using the spin-trap DMPO. 291
Dye photosensitization reactions in vitro may produce singlet oxygen or O2-, or both species, depending on the prevailing reaction conditions. In order to determine the identity of oxidizing species in vivo, it is important to 1) use in vitro model systems that mimic in vivo conditions as closely as possible, with regard to presence and concentration of targets, substrates, chelators, and trace metals; and 2)utilize in vivo methods of study as much as possible.
I would like to emphasize why we need to know, in as much detail as possible, about the identity of the oxidizing species and how they are produced. Without such information, our attempts to understand the biological role of these species and then to modify them are likely to fail. This is particularly important for strongly oxidizing species that act like the hydroxyl radical.
-H.M.Swartz297
Lethality due to oxygen radicals is an interesting phenomenon worthy of study; so is the production of these oxygen radicals by the photodynamic effect. This investigation is intended to shed light on the identity of cellular targets attacked by toxic oxygen species, on which of the oxygen species are involved in toxicity, and on factors involved in the photodynamic effect's production of oxygen species.
A major cellular target attacked by toxic oxygen species is likely to be DNA. Single-stranded nicking of DNA will be examined in this study in order to determine which factors are involved in this damage. Among the factors to be examined are the effects of scavengers of superoxide, hydrogen peroxide, and of hydroxyl free radical on the DNA damage; effectiveness of a wide range of dyes in classes including the xanthenes, acridines, thiazines, and a phenazine; effect of different amounts of iron and copper and of a wide range of biologically significant potential iron chelators; and the effects of a series of biological reductants on the production of DNA damage by the photodynamic effect.
The production of toxic oxygen species will be examined in this study by means of a series of assays. This study will use assays for oxidation of NADH and reduction of cytochrome c. It will examine the spectral shifts caused by intercalation into DNA of individual dyes to study intercalation, and the dyes' ability to reduce cytochrome c and oxidize NADH even when intercalated will be investigated. Production of hydroxyl radicals as assayed by two different assays will be examined in the presence of various scavengers of O2-, H2O2, and OH., as well as in the presence of different metal concentrations, different dyes, different chelators, and different reductants. Nicking of DNA will be examined similarly. Cellular levels of one biologically important reductant, glutathione, will be measured after exposure of the cells to dyes. Amelioration of kill levels will be determined for the presence of plasmids coding for catalase, SOD, and endonuclease IV, as well as preinduction of protective enzymes by PQ and Mn and the presence of thiourea. Finally, in vivo breakage of plasmid DNA will be looked at.
1. Bert, P. 1943. Barometric pressure: Researches in Experimental Physiology. Translated by M.A. Hitchcock and F. A. Hitchcock. College Book Company, Columbus, Ohio, p. 565-570.
2. Quebedeaux, B., U. D. Havelka, K. L. Livak, and R. W. F. Hardy. 1975. Effect of altered pO2 in the aerial part of soybean on symbiotic nitrogen fixation. Plant Physiol. 56:761-764.
3. Cooper, P. D., A. M. Burt, and J. N. Wilson.1958. Critical effect of oxygen tension on rate of growth of animal cells in continuous suspended culture. Nature 182:1508-1509.
4. Patz, A., A. Eastman, D. H. Higginbotham, and T. Klett. 1953. Oxygen studies in retrolental fibroplasia. Amer. J. Opthamol.36: 1511-1522.
5. Conger, A. D., and L. M. Fairchild. 1952. Breakage of chromosomes by oxygen. Proc. Natl. Acad. Sci. USA 38: 289-299.
6. Fenn, W. C., R. Gershman, and D. L. Gilbert. 1957. Mutagenic effects of high oxygen tensions on Escherichia coli. Proc. Natl. Acad. Sci. USA 43: 1027-1032.
7. Fuson, R.L., H.A. Saltzman, W.W.Smith, R.E. Whelan, S. Osterhout, R.T. Parker. 1965. Clinical hyperbaric oxygenation with severe oxygen toxicity. N. Engl. J. Med. 273:415-419.
8. Green, M. J., and H. A. O. Hill. 1984. Chemistry of Dioxygen. Meth. Enzymol. 105:3-22.
9. Gerschman, R., D.L. Gilbert, S.W. Nye, P. Dwyer, W.O. Fenn. 1954. Oxygen poisoning and X-irradiation: a mechanism in common. Science 119:623-626.
10. Fridovich, I. 1970. Quantitative aspects of the production of superoxide anion radical by xanthine oxidase. J. Biol. Chem. 245:4053-4057.
11. Nishikimi, M. 1975. Oxidation of ascorbic acid with superoxide anion generated by the xanthine/XO system. Biochem. Biophys. Res. Com. 63:463-468.
12. Knowles, P. F., J. F. Gibson, F. M. Pick, and R. C. Bray. 1969. Electron-spin-resonance evidence for enzymic reduction of oxygen to a free radical, the superoxide ion. Biochem. J. 111: 53-58.
13. Misra, H.P., and I. Fridovich. 1972. The univalent reduction of oxygen by reduced flavins and quinones. J. Biol. Chem. 247: 188-192.
14. Bartoli, G., T. Galeotti, and A. Azzi. 1977. Production of superoxide anions and hydrogen peroxide in ehrlich ascites tumour cell nuclei. Biochim. Biophys. Acta 497: 622-626.
15. Patton, S.E., G.M. Rosen, and E.J. Raukman. 1980. Superoxide production by purified hamster hepatic nuclei. Mol. Pharmacol. 18: 588-593.
16. Asada, K., and K. Kiso. 1973.The photo-oxidation of epinephrine by spinich chloroplasts and its inhibition by SOD: evidence for the formation of superoxide radicals in chloroplasts. Agr. Biol. Chem. 27: 453-457.
17. Kono, Y., M.-A. Takahashi, and K. Asada. 1976. Oxidation of manganous pyrophosphate by superoxide radicals and illuminated spinich chloroplasts. Arch. Biochem. Biophys. 174: 454-462.
18. Bovaris, A., R.A.Sanchez, and M.T. Beconi. 1978. Antimycin and cyanide-resistant respiration and superoxide anion production in fresh and aged potato tuber mitochondria. FEBS Letters 92: 333-338.
19. Johnston, R.B., Jr., J.E. Lehmeyer, and L.A. Guthrie. 1976. Generation of superoxide anion and chemiluminescence by human monocytes during phagocytosis and on contact with surface-bound immunoglobulin G. J. Exp. Med. 143: 1551-1556.
20. Johnston, R.B., Jr., C.A.Godzik and Z.A. Cohn. 1978. Increased superoxide anion production by immunologically activated and chemically elicited macrophages. J. Exp. Med. 148: 115-127.
21. Aust, S.D., D.L. Roerig, and T.C. Pederson. 1972. Evidence for superoxide generation by NADPH-cytochrome c reductase of rat liver microsomes. Biochem. Biophys. Res. Com. 47: 1133-1136.
22. Simon, R.H., C.H. Scoggin, and D. Patterson. 1981. Hydrogen peroxide causes the fatal injury to human fibroblasts exposed to oxygen radicals. J. Biol. Chem. 256: 7181-7186.
23. Misra, H.P., and I. Fridovich. 1972. The generation of superoxide radical during the autoxidation of hemoglobin. J. Biol. Chem. 247: 6960-6962.
24. Gotoh, T., and K. Shikama. 1976. Generation of the superoxide radical during the autoxidation of oxymyoglobin. J. Biochem. 80: 397-399.
25. Misra, H.P., and I. Fridovich. 1971. The generation of superoxide radical during the autoxidation of ferredoxins. J. Biol. Chem. 246: 6886-6890.
26. Ballou, D., G.Palmer, and V. Massey. 1969. Direct demonstration of superoxide anion production during the oxidation of reduced flavin and its catalytic decomposition by erythrocuprein. Biochem. Biophys. Res. Com. 36: 898-904.
27. McCord, J.M., and I. Fridovich. 1973. Production of superoxide anion in photolyzed water demonstrated through the use of superoxide dismutase. Photochem. Photobiol. 17: 115-121.
28. Carmichael, A.J., A. Samuni, and P. Rietsz. 1985. Photogeneration of superoxide and decarboxylated peptide radicals by carboquone, mitomycin C and streptonigrin. Photochem. Photobiol. 41: 635-641.
29. Hassan, H.M., and I. Fridovich. 1978. Superoxide radical and the oxygen enhancement of the toxicity of paraquat in Escherichia coli. J. Biol. Chem. 253: 8143-8148.
30. Bielski, B.H.J., and H.W. Richter. 1977. A study of the superoxide radical chemistry by stopped-flow radiolysis and radiation induced oxygen consumption. J. Amer. Chem. Soc. 99: 3019-3023.
31. Nishikimi, M., H. Yamada, and K. Yagi. 1980. Oxidation by superoxide of tocopherols dispersed in aqueous media with deoxycholate. Biochim. Biophys. Acta 627: 101-108.
32. Bielski, B.H.J., and P.C.Chan. 1975. Kinetic study by pulse radiolysis of the lactate dehydrogenase-catalyzed chain oxidation of nicotinamide adenine dinucleotide by HO2. and O2- radicals. J. Biol. Chem. 250: 318-321.
33. Sawyer, D.T., and J.S. Valentine. 1981. How super is superoxide?Acc. Chem. Res. 14: 393-400.
34. Kono, Y., M.-A. Takahashi, and K. Asada. 1976. Oxidation of manganous pyrophosphate by superoxide radicals and illuminated spinich chloroplasts. Arch. Biochim. Biophys. 174: 454-462.
35. Butler, J., and B. Halliwell. 1982. Reaction of iron-EDTA chelates with the superoxide radical. Arch. Biochem. Biophys. 218:174-178.
36. McCord, J. M., and I. Fridovich. 1968. The reduction of cytochrome c by milk xanthine oxidase. J. Biol. Chem. 243: 5753-5760.
37. Bielski, B.H.J., G.G. Shiue, and S. Bajuk. 1980. Reduction of Nitro Blue Tetrazolium by CO2- and O2- Radicals. J. Phys. Chem. 84: 830-832.
38. Bielski, B.H.J., R.L.Arudi, and M.W. Sutherland. 1983. A study of the reac tivity of HO2/O2- with unsaturated fatty acids. J. Biol. Chem. 258: 4759-4761.
39. Misra, H.P. 1974. Generation of superoxide free radical during the autoxidation of thiols. J. Biol. Chem. 249: 2151-2155.
40. Marklund, S., and G. Marklund. 1974. Involvement of the superoxicde anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur. J. Biochem. 47:469-474.
41. Misra, H.P., and I. Fridovich. 1972. The role of superoxide anion in the auto xidation of epinephrine and a simple assay for superoxide dismutases. J. Biol. Chem. 247:3170-3175.
42. Kono, Y., and I. Fridovich. 1982. Superoxide radical inhibits catalase. J. Biol. Chem. 257: 5751-5754.
43. Blum, J., and I. Fridovich. 1984. Inactivation of the selenium-glutathione peroxidase by superoxide. Fed. Proc. 43: 2011-2015.
44. Halliwell, B. and J. M. C. Gutteridge. 1985. Free Radicals in Biology and Medicine. Clarendon Press, Oxford, p. 280.
45. McCord, J.M. 1974. Free radicals and inflammation: protection of sinovial fluid by superoxide dismutase. Science 185: 529-531.
46. Lynch, R.E., and I. Fridovich. 1978. Effects of superoxide on the erythrocyte membrane. J. Biol. Chem. 253:1838-1845.
47. Rosen, G. M., M. J. Barber, and E. J. Raukman. 1983. Disruption of erythrocyte membranal organization by superoxide dismutase. J. Biol. Chem. 258: 2225-2228.
48. Girotti, A.W. 1985. Lipid photoxidation in erythrocyte ghosts-sensitization of the membranes toward ascorbate-induced and superoxide-induced peroxidation and lysis. Arch. Biochem. 236: 238-251.
49. Brawn, K., and I. Fridovich. 1981. DNA Strand Scission by enzymically generated oxygen radicals. Arch. Biochem. Biophys. 206: 414-419.
50. Lesko, S.A., R.J. Lorentzen, and P.O.P. Ts'o. 1980. Role of superoxide in DNA strand scission. Biochemistry 19: 3023-3028.
51. Ross, W.E., E.R. Block, and R.Y. Chang. 1979. Paraquat-induced DNA damage in mammalian cells. Biochem. Biophys. Res. Comm. 91:1302-1308.
52. Babior, B., J.T. Curnutte, and R.S. Kipnes. 1975. Biological defense mechanisms: evidence for the participation of superoxide in bacterial killing by xanthine oxidase. J. Lab. Clin. Med. 85: 235-244.
53. Cunningham, M.L., and B.R. Lokesh. 1983. Superoxide anion generated by KO2 is cytotoxic and mutagenic to Chinese hamster ovary cells. Mutat. Res. 121: 299-304.
54. Weitzman, S.A., A.B. Weitberg, E. Clark, and T.Stossel. 1985. Phagocytes as carcinogens: malignant transformation produced by human neutrophils. Science 227: 1231-1233.
55. Weitzman, S.A., A.B. Weitberg, E. Clark, and T.Stossel. 1985. Phagocytes as carcinogens: malignant transformation produced by human neutrophils. Science 227: 1231-1233.
56. Fee, J.A. 1980. Is superoxide toxic?, p.41-48. In W.H. Bannister and J.V. Bannister (eds.), Biological and Clinical Aspects of Superoxide and Superoxide Dismutase. Elsevier/North Holland, New York.
57. Baum, R.M. 1984. Superoxide theory of oxygen toxicity is center of heated debate. Chemical and Engineering News April 9, 1984: p. 20-26.
58. Fee, J.A. 1981. Is superoxide toxic and are superoxide dismutases essential for aerobic life? In M.A.J. Rodgers and E.L. Powers (eds.), Oxygen and Oxyradicals in Chemistry and Biology. Academic Press, New York.
59. J.S. Valentine. 1979. The Chemical Reactivity of Superoxide Anion. In W.S. Caughey (ed.), Biochemical and Clinical Aspects of Oxygen. Academic Press, New York.
60. Bielski, B.H.J., and G. Schive. 1979. Reaction rates of superoxide radical with the essential amino acids, p. 43-65. In D. W. Fitzsimons (ed.), Oxygen Free Radicals in Tissue damage. Elsevier/North-Holland, Amsterdam.
61. Loschen, G., A. Azzi, C. Richler, and L. Flohe. 1974. Superoxide radicals as precursors of mitochondrial hydrogen peroxide. FEBS Letters 42: 68-72.
62. Turrens, J.F., Freeman, B.A., and Crapo, J.D. 1982. Hyperoxia increases hydrogen peroxide release by lung mitochondria and microsomes. Arch. Biochem. Biophys. 217: 411-421.
63. Cohen, G., and Heikkila, R. 1974. The generation of hydrogen peroxide, su peroxide radical and hydroxyl radical by 6-hydroxydopamine, dialuric acid and related cytotoxic agents. J. Biol. Chem. 249:2447-2452.
64. May, J.M., and C. de Ha�n. 1979. Insulin-stimulated intracellular hydrogen peroxide production in rat epididymal fat cells. J. Biol. Chem. 254: 2214-2220.
65. Zigler, J.S., Jr., H.M. Jernigan, Jr., D. Garland, and V.N. Reddy. 1985. The effects of "oxygen radicals" generated in the medium on lenses in organ culture: inhibition of damage by chelated iron. Arch. Biochem. Biophys. 241: 163-172.
66. Uchida, Y., H. Shigematu, and K. Yamafuji. 1965. The mode of action of hydrogen peroxide on DNA. Enzymologia 6: 369-376.
67. Keller, K.M., and E.C.Pollard. 1977. Action of hydrogen peroxide on degradation of DNA after irradiation in E.coli. Int. J. Radiat. Biol. 31: 407-413.
68. Massie, Y., M. Kitahara, K. Maeda, and H. Umezawa. 1972. The kinetics of degradation of DNA and RNA by hydrogen peroxide. Bioc. Biop. Acta 272: 539-548.
69. Meneghini, R., and M.E. Hoffmann. 1980. The damaging action of hydrogen peroxide on DNA of human fibroblasts is mediated by a non-dialyzable compound. Biochim. Biophys. Acta 608: 167-173.
70. McCormick, J.P., J.R. Fischer, J.P. Pachlatki, and A.A. Eisenstark. 1976. Characterization of a cell-lethal product from the photooxidation of tryptophan: hydrogen peroxide. Science 191: 468-469.
71. Plaine, H.L. 1955. The effect of oxygen and of hydrogen peroxide on the action of a specific gene and on tumor induction in Drosophila melanogaster. Genetics 40: 268-280.
72. Bray, R.C., and L. Calabres. 1974. Reduction and inactivation of superoxide dismutase by hydrogen peroxide. Biochem.J. 139: 43-47.
73. F. Haber and J. Weiss. 1934. The catalytic decomposition of hydrogen peroxide by iron salts. Proc. Roy. Soc. London (A) 147:332-342.
74. Koppenol, W.H., J. Butler, and J.W. van Leeuwen. 1978. The Haber-Weiss cycle. Photochem. Photobiol. 28:655-660.
75. Czapski, G. 1984. Reaction of hydroxyl radical. Meth. Enzymol. 105: 209-215.
76. Halliwell, B., and J.M.C. Gutteridge. 1984. Role of iron in oxygen radical reactions. Meth. Enzymol. 105: 47-56.
77. Anonymous. 1970. USDA handbook of nutritional values in foods. Dover Pub lications, New York.
78. Buettner, G.R., L.W. Oberley, and S.W. Leuthaus. 1978. The effect of iron on the distribution of superoxide and hydroxyl radicals as seen by spin trapping and on the superoxide dismutase assay. Photochem. Photobiol. 28:693-695.
79. Halliwell, B. 1978.Superoxide-dependent formation of hydroxyl radicals in the presence of iron salts-its role in degradation of hyaluronic acid by a superoxide-generating system. FEBS Letters 96: 238-242.
80. Mello-Filho, A.C., and R. Meneghini. 1984. In vivo formation of single-strand breaks in DNA by hydrogen peroxide is mediated by the Haber-Weiss reaction. Biochem. Biophys. Acta 781: 56-63.
81. Sutton, H.C., and C.C. Winterbourne. 1984. Role of Metal Chelating Agents as Catalysts in a Hydroxyl Radical Forming Process- a comparison with the Haber-Weiss reaction, p.177-183. In W. Bors (ed.), Oxygen Radicals in Chemistry and Biology. Walter de Gruyter & Co, Berlin.
82. Buettner, G.R., L.W.Oberley, and S.Chan Leuthauser. 1978. The effect of iron on the distribution of superoxide and hydroxyl radicals as seen by spin trapping and on the superoxide dismutase assay. Photochem. Photobiol. 28: 693-697.
83. Floyd, R.A. 1981. DNA-ferrous iron catalyzed hydroxyl free radical formation from hydrogen peroxide. Biochem. Biophys. Res. Com. 99: 1209-1215.
84. Motohashi, N., and I. Mori. 1983. Superoxide-dependent formation of the hydroxyl radical catalyzed by transferrin. FEBS Letters 157: 197-199.
85. Gutteridge, J.M.C., S.K. Paterson, A.W. Segal, and B. Halliwell. 1981. Inhibition of lipid peroxidation by the iron-binding protein lactoferrin. Biochem. J. 199: 259-261.
86. Richmond, R. and B. Halliwell. 1982. Formation of hydroxyl free radical from the paraquat radical cation, demonstrated by a highly specific GC technique. The role of superoxide, hydrogen peroxide, and glutamine reductase. J. Inorg. Biochem. 17: 95-107.
87. Buettner, G.R., and L.W. Oberley. 1979. The production of hydroxyl radical by tallysomycin and copper(II). FEBS Letters 101: 333-335.
88. Winterbourn, C.C. 1981. Evidence for the production of hydroxyl radicals from the adriamycin semiquinone and hydrogen peroxide. FEBS Letters 136: 89-94.
89. Lai, C.-S., and L.H. Piette. 1977. Hydroxyl radical production involved in lipid peroxidation of rat liver microsomes. Biochem. Biophys. Res. Com. 78: 51-59.
90. Tauber, A.I., and B.M. Babior. 1977. Evidence for hydroxyl radical production by human neutrophils. J. Clin. Invest. 60: 374-379.
91. Weiss, S.J., G.W. King, and A. F. LoBuglio. 1977. Evidence for hydroxyl radical generation by human monocytes. J. Clin. Invest. 60: 370-373.
92. Dorfman, L.M., and G.E. Adams. 1973. Reactivity of the Hydroxyl Radical in Aqueous Solutions. National Standard Reference Data Service- National Bureau of Standards No. C13.48:46.
93. Lesko, S.A., J.-L. Drocourt, and S.-U. Yang. 1982. DNA-protein and DNA-interstrand cross-links induced in isolated chromatin by hydrogen peroxide and ferrous EDTA chelates. Biochemistry 21: 5010-5015.
94. Schellenberg, K.A. 1979. Reaction of hydroxyl radical with thymidine. Scavenging by a dihydropyridine. Fed. Proc. Abstr. 38: 501.
95. Rosen, H., and S. J. Klebanoff. 1981. Role of iron and EDTA in the bactericidal activity of a superoxide anion-generating system. Arch. Biochem. Biophys. 208: 512-519.
96. Heikkila, R.E., B. Winston, G. Cohen, and H. Barden. 1976. Alloxan-induced diabetes. Evidence for hydroxyl radical as a cytotoxic intermediate. Biochem. Pharm. 25: 1085-1092.
97. Grankvist, K., S. Marklund, J. Sehlin, and I. Taljedal. 1979. Influence of trace metals on alloxan cytotoxicity in pancreatic islets. FEBS Letters 105: 15-18.
98. Kutsuki, H., and M.H. Gold. 1982.Generation of hydroxyl radical and its involvement in lignin degradation by Phanerochaete chrysosporium. Biochem. Biophys. Res. Com.109: 320-325.
99. Percival, M.P., and A.D.Dodge. 1983. Photodynamic effects of rose bengal on senescent flax cotyledons. J. Exp. Biol. 34(138)47-54.
100. Wasserman, H.H., and R.W. Murray, eds. 1978. Singlet Oxygen. New York: Academic Press.
101. Ranby, B., and J.F. Rabek, eds. 1978. Singlet oxygen reactions with organ ic compounds and polymers. New York: Wiley.
102. Fridovich, I. 1974. Superoxide dismutases. Advances in Enzymology 41:35-97.
103. R.A. Weisiger and I. Fridovich, J. Biol. Chem.248:3582-3592(1973) Superoxide dismutase: organelle specificity.
104. McCord, J.M. and I. Fridovich. 1969. Superoxide dismutase: an enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 244:6049-6055.
105. Keele, B.B., Jr., J.M. McCord, and I. Fridovich. 1971. Further characteriza tion of bovine superoxide dismutase and its isolation from bovine heart. J. Biol. Chem. 246:2875-2880.
106. Carrico, R.J., and H.F. Deutsch. 1970. The presence of zinc in human cy tocuprein and some properties of the apoprotein. J. Biol. Chem. 245:723-727.
107. Harris, J.I., and H.M. Steinman. 1977. Amino acid sequence homologies among superoxide dismutases. In "Superoxide and Superoxide Dismu tases." A.M.Mitchellson, J.M.McCord, and I. Fridovich, eds., Academic Press, London. pp225-230.
108. Pugh, S.Y.R., and I. Fridovich. 1985. Induction of superoxide dismutases in Escherichia coli B by metal chelators. J. Bacteriol. 162:196-202.
109. Hassan, H.M., and I. Fridovich. 1979. Intracellular production of superoxide radical and of hydrogen peroxide by redox active compounds. Arch. Bio chem. Biophys. 196:385-395.
110. Claibourne, A., and Fridovich, I. 1979. Purification of the o-dianisidine peroxidase of Escherichia coli B. J. Biol. Chem. 254:4245-4252.
111. Loewen, P.C., B.L. Triggs, C.S. George, and B.E. Hrabarchuk. 1985. Genetic mapping of katG, a locus that affects synthesis of the bifunctional catalase-peroxidase hydroperoxidase I in Escherichia coli. J. Bacteriol. 162:661-667.
112. Schonbaum, G.R., and B. Chance. 1976. Catalase. In The Enzymes, vol. XIII, P.D. Boyer, ed. Academic Press, New York.
113. Richter, H.E., and P.C. Loewen. 1981. Induction of catalase in Escherichia coli by ascorbic acid involves hydrogen peroxide. Biochem. Biophys. Res. Comm. 100:1039-1046.
114. Hassan, H.M. and I Fridovich. 1978. Regulation of the synthesis of cata lase and peroxidase in Escherichia coli. J. Biol. Chem. 253:6445-6450.
115. Tuveson, R.W., and M.E.March. 1980. Photodynamic and sunlight inactiva tion of Escherichia coli strains differing in near-uv sensitivity and recom bination proficiency. Photochem. Photobiol.31:287-289.
116. Loewen, P.C., B.L. Triggs, G.R. Klassen, and J.H. Weiner. 1983. Identification and physical characterization of a col E1 hybrid plasmid containing a catalase gene of Escherichia coli . Can. J. Biochem. Cell Biol. 61:1315-1321.
117. Martin, J.P., Jr. Unpublished observation, 1986.
118. Christman, M.F., R.W. Morgan, F.S. Jacobson, and B.N. Ames. 1985. Positive control of a regulon for defenses against oxidative stress and some heat-shock proteins in Salmonella typhimurium. Cell 41: 753-762.
119. Triggs-Raine, B.L., and P.C. Loewen. 1987. Physical characterization of katG, encoding HPI of Escherichia coli. Gene 52:121-128.
120. Nettleton, C.J., C. Bull, T.O. Baldwin, and J.A.Fee. 1984. Isolation of the Escherichia coli iron superoxide dismutase gene: Evidence that intracellular superoxide concentration does not regulate oxygen-dependent synthesis of the mangenese superoxide dismutase. Proc. Natl. Acad. Sci. USA 81:4970-4973.
121. Sakamoto, H., and D. Touati. 1984. Cloning of the iron superoxide dismutase gene (sodB ) in Escherichia coli K-12. J. Bacteriol. 159:418-420.
122. Touati, D. 1983. Cloning and mapping of the manganese superoxide dis mutase gene (sodA) of Escherichia coli K-12. J. Bacteriol. 155:1078-1087.
123. Loeb, L.A. and B.D. Preston. 1986. Mutagenesis by apurinic/apyrimidinic sites. Ann. Rev. Genet. 20:201-30.
124. Duncan, B.K. 1981. DNA glycosylases. In The Enzymes, ed. P.D. Boyer, 14:565-86. New York: Academic Press.
125. Henner, W.D., S.M. Grunberg, and W.A. Haseltine. 1983. Enzyme action at 3' termini of ionizing radiation-induced DNA strand breaks. J. Biol. Chem. 258(24):15198-15205.
126. Denq, R.-Y. and I. Fridovich. 1987. Formation of thymine glycol as a con sequence of oxygen radical attack on DNA, in vitro and in vivo. Submitted to J. Biol. Chem. Aug 1987.
127. Demple, B., A. Johnson, and D. Fung. 1986. Exonuclease III and endonu clease IV remove 3' blocks from DNA synthesis primers in H2O2-damaged Escherichia coli. Proc. Natl. Acad. Sci. USA 83:7731-7735.
128. Walker, G.C., L. Marsh, and L.A. Dodson. 1985. Genetic analyses of DNA repair: inference and extrapolation. Ann. Rev. Genet. 19:103-26.
129. Janovska, E., V. Zhestjanikov, and M. Vizdalova. 1970. On the nature of reparable and non-reparable lethal damage in E. coli and bacteriophage induced by the photodynamic action of acridine orange. Int. J. Radiat. Biol. 18(4):317-329.
130. Freifelder, D. 1987. Molecular Biology, second edition, chapter 10. Jones & Bartlett Publishers, Inc., Boston.
131. Brawn, M.K., and I. Fridovich. 1985. Increased superoxide radical produc tion evokes inducible DNA repair in Escherichia coli. J. Biol. Chem. 260:922-925.
132. Harrison, A.P., J.L. Hafner, and C. O'Brien. 1972. Ultraviolet-killing and photodynamic-killing in mutants of Escherichia coli. Mutation Res., 14:447-449.
133. Chan, E., and B. Weiss. 1987. Endonuclease IV of Escherichia coli is in duced by paraquat. Proc. Natl. Acad. Sci. USA 84:3189-3193.
134. Sancar, A., and G.B. Sancar. 1988. DNA repair enzymes. Ann. Rev. Bio chem. 57:29-67.
135. Cunningham, R.P., S.M. Saporito, S.G. Spitzer, and B. Weiss. 1986. Endo nuclease IV (nfo) mutant of Escherichia coli. J. Bact. 168:1120-1127.
136. Fridovich, I. 1986. Biological effects of the superoxide radical. Arch. Bio chem. Biophys. 247:1-11.
137. Blum, H.F. 1941. Photodynamic Action and Diseases Caused by Light. Reinhold, New York.
138. Morliere, P. 1986. Drug-induced photosensitiviy: phototoxic and photoal lergic reactions-a few molecular aspects. Biochimie 68(6):849-856.
139. Foote, C.S. 1976. Photosensitized oxidation and singlet oxygen: conse quences in biological systems. In: Free radicals in Biology, volume II, Wil liam A. Pryor, ed. Academic Press, New York.
140. Crow, K.D., E. Alexander, W.H.L. Buck, B.E. Johnson, and I.A. Magnus. 1961. Photosensitivity due to pitch. Br. J. Derm. 73:220-232.
141. Heitz, J.R., and K. R. Downum, eds. 1987. Light-activated pesticides. Washington, D.C.: American Chemical Society.
142. Chacon, J.N., J. McLearie, and R.S. Sinclair. 1988. Singlet oxygen yields and radical contributions in the dye-sensitized photoxidation in methanol of esters of polyunsaturated fatty acids (oleic, linoleic, linolenic and arachi donic).Photochem. Photobiol. 47(5)647-656.
143. Odell, G.B., R. Schaffer, and A.P. Simonpoulos, eds. 1974. Phototherapy in the newborn: an overview. Washington, D.C.: National Academy of Sci ence USA.
144. Felber, T.D., E.B. Smith, J.M. Knox, C. Wallis, and J.L. Melnick. 1973. Pho todynamic inactivation of Herpes simplex. J. Amer. Med. Assoc. 223:289-292.
145. Barnhart, E.R. (ed. ). 1986. Physicians's Desk Reference. Oradell, New Jersey: Medical Economics Company.
146. Ito, T., and K. Kobayashi. 1977. A survey of in vivo photodynamic activity of xanthenes, thiazines, and acridines in yeast cells. Photochem. Photo biol. 26:581-587.
147. Blazek, E.R., and P.V. Hariharan. 1984. Alkaline elution studies of hemato porphorin-derivative photosensitized DNA damage and repair in chinese hamster ovary cells. Photochem. Photobiol. 40(1):5-13.
148. Wagner, Stephen, W.D. Taylor, A. Keith, and W. Snipes. 1980. Effects of acridine plus near ultraviolet light on E coli membranes and DNA in vivo. Photochem. Photobiol. 32:771-779.
149. Bellin, JS., and L.I. Grossman. 1965. Photodynamic degradation of nucleic acids. Photochem. Photobiol. 4:45-53.
150. Grossweiner, L.I., and A.G. Kepka. 1972. Photosensitization in biopolym ers. Photochem.Photobiol. 16: 305-314.
151. Spikes, J.D., and M.L. MacKnight. 1970. Dye-sensitized photoxidation of proteins. Annals New York Acad. Sci. 171:149-162.
152. Brault, D., C. Veverbiz, and M. Dellinger. 1986. Fundamental aspects in tumor photochemotherapy: interactions of porphyrins with membrane mod el systems and cells.Biochimie 68(6):913-922.
153. Gutter, B, W.T. Speck, and H.S. Rosenkrantz. 1977. Light-induced muta genicity of neutral red (3-amino-7-dimethyl-2-methylphenazine hydro chloride). Cancer Res. 37:1112-1114.
154. Barltrop, J., B,B, Martin, and D.F. Martin. 1983. Ptychodiscus brevis as a model system for photodynamic action. Microbios 37:95-103
155. Lamberg, S.I. 1967. A new photosensitizer, the artificial sweetener cycla mate. J. Amer. Med. Assoc. 201:747-750.
156. Harber, L.C., D.P. Torres, A.W. Kopf, A.A. Fisher, and L. Frank. 1964. Ber loque Dermatitis. Arch. Dermatol. 90:572-576.
157. Wilkenson, D.S. 1961. Photodermatitis due to tetrachlorsalicylanilide. Br. J. Dermatol. 73:213-219.
158. Birmingham, D.J., M.M.Key, and G.E. Tubich. 1961. Phototoxic Bullae among celery harvesters. Arch. Dermatol. 83:73-87
159. Seely, T. 1977. Mechanisms of the photosensitized oxidation of tyramine. Photochem. Photobiol.26:115.
160. Epstein, J.H., L.A. Brunestine, M.C. Petrsen, and B.E. Schwarz. 1957. A study of photosensitivity occurring with chlorpromazine therapy. J. Invest. Dermatol. 28:329-338.
161. Epstein, S. 1968. Chlorpromazine photosensitivity. Arch. Dermatol. 98:354-363.
162. Morris, W.E. 1960. Photosensitivity due to tetracycline derivative. J. Amer. Med. Assoc. 172:1155-1156.
163. Falk, M.S. 1960. Light sensitivity due to demethychlortetracycline. J. Amer. Med. Assoc. 172:1156-1157.
164. Harber, L.C., T.A. Tromovitsh, and R.L.Baer. 1961. Studies on photosensi tivity due to demethylchlortetracycline. J. Invest. Dermatol. 37:189-193.
165. Peterkin, G.A. 1945. Skin Eruptions due to the local application of sulpha nilamides. Br.J. Dermatol.57:1-9.
166. Blum, H.F. 1941. Studies of photosensitivity due to sulfanilamide. J. Invest. Dermatol. 4:159-173.
167. Epstein S. 1939. Photoallergy and primary photosensitivity to sulfanil amide. J. Invest. Dermatol.2:43-51.
168. DeCuyper, J., J.Piette, and A. Van de Vorst. 1983. Activated oxygen spe cies produced by photoexcited furocoumarin derivatives. Arch. Int. Physiol. Biochim.91:471-476.
169. Stevanovic, D.V. Photosensitivity due to certain drugs. Br. J. Derm. 73:233-237.
170. Bakri, A., G.M. Vanheneg, and J.L. Chanal.1983. Photopharmacology of the tranquilizer chlordiaxepoxide in relation to its phototoxicity. Photochem. Photobiol. 38:177-183.
171. Ramsey, C.A., and E.Obreshkova. 1974. Photosensitivity from nalidixic acid. Br. J. Dermatol. 91:523-528.
172. Chahidi, C., P. Morliere, M. Aubailly, L. Dubertret, and R. Santus. 1983. Photosensitization by methotrexate photoproducets. Photochem. Photo biol. 38:317-322.
173. Reszke, K., and C.F.Chignell. 1983. Spectroscopic studies of cutaneous photosensitizing agents-IV. The photolysis of benoxaprofen, an anti-in flammatory drug with phototoxic properties. Photochem. Photobiol. 38:281-291
174. Harber, L.C., I.E. Kochevar, and A.R. Shalita. 1982. In The Science of Pho tomedicine (Regan J.D. & Parrish J.A., eds.), Plenum Press, New York pp323-347.
175. Beani, J.-C., P. Amblerd, and J.-L. Reymond. 1986. Problemes poses par l'utilisation therapeutique des photosensibilisateurs en dermatologie. Bio chimie 68(6):905-912.
176. Regan, J.D., and R.B. Setlow. 1977. The effect of proflavin plus visible light on the DNA of human cells. Photochem. Photobiol. 25:345-346.
177. Jarratt, M. 1977. Photodynamic inactivation of herpes simplex virus. Photo chem. Photobiol. 25:339-340.
178. Oxman, M. 1977. The clinical evaluation of phtotodynamic inactivation for the therapy of recurrent herpes simplex virus infections.Photochem. Photo biol. 25:343-344.
179. Patrice, T., V. Praloran, M. Lebodic, and L. Lebodic. 1986. Experimental aspects of in vitro and in vivo photochemotherapy. Biochimie 68(6):923-926.
180. Dougherty, T., and T.W. Sery. 1985. Photoradiation of rabbit ocular malig nant melanoma sensitized with hematoporphyrin derivative. Curr. Eye Res. 3(4):519-528.
181. Houba-Herrin, N. 1982. Mechanisms for dye mediated photodynamic ac tion: singlet oxygen production, deoxygianosine oxidation, and phage inac tivating efficiencies. Photochem. Photobiol.36:297-306.
182. Linen, S.M., and D.C. Neckers. 1988. Type I and Type II sensitizers based on rose bengal onium salts. Photochem. Photobiol. 47(4):543-550.
183. Martin, J.P., and N.J. Logsden. 1987. Oxygen radicals mediate cell inac tivation by acridine dyes, fluorescein, and lucifer yellow CH. Photochem. Photobiol. 46:45-53.
184. Nikaido, H., and E.Y. Rosenberg. 1983. Porin channels in Escherichia coli: Studies with Liposomes reconstituted from purified proteins. J. Bacteriol. 153:241-252.
185. Nikaido, H., E.Y. Rosenberg, and J. Foulds. 1983. Porin channels in Escheri chia coli: Studies with �-lactams in intact cells. J. Bacteriol. 153:253-257.
186. Bellin, J.S., L. Lutwick, and B. Jonas. 1969. Effects of photodynamic action on E. coli. Arch. Biochem. Biophys. 132:157-164.
187. Hassan, H.M., and I. Fridovich. 1980. Mechanism of the antibiotic action of pyocyanine. J. Bacteriol. 141:156-163.
188. Berns, M.W., R.S. Olson, and D.E.Rounds. 1968. In vitro production of chromosomal lesions with an argon laser microbeam. Nature 221:74-75.
189. Berns, M.W., R.S. Olson, and D.E.Rounds. 1969. Effects of laser micro- irradiation of chromosomes. Exp. Cell Res. 56:297-298.
190. Windholz, M. (ed.). 1983. The Merck Index, tenth edition. Merck, Rahway, New Jersey.
191. Paczkowska, B., J. Paczkowska, and D.C. Neckers. 1985. Properties of Rose bengal:(X) new derivatives of polymer-rose bengal. Photochem. Photobiol. 42(5):603-604.
192. Anonymous. 1988. Code of Federal Regulations, Title 21. Washington: U.S. Government Printing Office.
193. Beuttner, G.R., T.P. Doherty, and T.D. Bannister. 1984. Hydrogen peroxide and hydroxyl radical formation by methylene blue in the presence of ascor bic acid. Radiat. Environ. Biophys. 23:235-243.
194. Welsh, J.N., and M.H. Adams. 1954. Photodynamic inactivation of bacte riophage. J. Bacteriol. 68:122-127.
195. Yamamoto, N. 1958. Photodynamic inactivation of bacteriophage and its inhibition. J. Bacteriol. 75:443-478.
196. Kaufman, E. and C.W. Hiatt. 1959. Photodynamic action of proflavin on T2 colipage. Virology 9:478-479.
197. Wallis, C., and J.L. Melnick. 1965. Photodynamic inactivation of animal vi ruses: a review. Photochem. Photobiol. 4:159-170.
198. Bezman, S.A., P.A. Burtis, T.P.J. Izod, and M.A. Thayer. 1978. Photody namic inactivation of E. coli by rose bengal immobilized on polystryrene beads. Photochem. Photobiol. 28:325-330.
199. Wakayama, Y., Takagi, M., and Yano, K. 1980. Photosensitized inactivation of E. coli cells in toluidine blue-light system. Photochem. Photobiol. 32:601-605.
200. Martin, J.P., and N. Logsdon. 1987. The role of oxygen radicals in dye-me diated photodynamic effects in Escherichia coli B. J. Biol. Chem. 262:7213-7219.
201. Cooney, J.J., and N.I. Krinsky.1972. Photodynamic killing of Acholeplasma laidlawii. Photochem. Photobiol. 16:523-526.
202. Schwartzel, E.H. and J.J. Cooney. 1974. Action of light on Micrococcus roseus. Can. J. Microbiol. 20:1015-1021.
203. Ito, A., and T. Ito. 1982. Enhancing effect of ascorbate on toluidine blue- photosensitization of yeast cells. Photochem. Photobiol.35:501-505.
204. Morgan, D.D., and D. Warshawsky. 1977. The photodynamic immobiliza tion of Artemia salina nauplii by polycyclic aromatic hydrocarbons abd its relationship to carcinogenic activity. Photochem Photobiol. 25:39-46.
205. Pooler, J. 1972. Photodynamic alteration of sodium currents in lobster ax ons. J. Gen. Phys. 60:367-387.
206. Miller, J.P. and A.I. Selverston. 1979. Rapid killing of single neurons by ir radiation of intracellularly injected dye. Science 206:702-704.
207. Stewart, W.W. 1978. Functional coupling between cells as revealed by dye coupling with a highly fluorescent napthalimide tracer. Cell 14:741-759.
208. Stark, W.S., and S.D. Carlson. 1984. Blue and ultraviolet light induced damage to the drosophila retina: ultrastructure. Curr. Eye Res. 3(12):1441-1454.
209. Stark, W.S., K.D.Walker, and J.M. Eidel. 1985. Blue and ultraviolet light in duced damage to the drosophila retina:microspectrophotometry and elec trophysiology. Curr. Eye Res. 4(10):1059-1075.
210. Percival, M.P., and A.D.Dodge. 1983. Photodynamic effects of rose bengal on senescent flax cotyledons. J. Exp.Biol. 34(138)47-54.
211. Foote, C.S. 1970. Quenching of singlet oxygen. Ann. N. Y. Acad. Sci. 171:139-148.
212. Peyron, J.-L., and J. Meynadier. 1986. The pharmacological basis for the treatment of photodermatoses. Biochimie 68(6):899-904.
213. Salet, C. 1986. Hematoporphyrin and hematoporphyrin-derivative photo sensitization of mitochondria. Biochimie 68(6):865-868.
214. Haga, J.Y., and J.D. Spikes. 1972. Effects of Photodynamic Treatment. In Research Progress in Organic, Biological and Medical Chemistry, vol III, pp 464-479. Gallo, U., and L. Santamaria (eds). North-Holland, Amster dam.
215. Garuin, R.T., G.R. Julian, and S.J. Rogers. 1969. Dye-sensitized photoox idation of the Escherichia coli ribosome. Science 164:583-584.
216. Salzberg, B.M. 1983. Optical recording of Electrical Activity in Neurons Us ing Molecular Probes. In Current Methods in Cellular Neurobiology,pp 177-185. J. Barker & J. McKelvy (eds.) John Wiley & Sons, New York.
217. Gurnani, S., M.Arifuddin, and K.T. Augusti. 1966.Effects of light on amino acids-I. Tryptophan. Photochem. Photobiol. 5:495-505.
218. Friefelder, D., PF. Davison, and E.P. Geiduschek. 1961. Damage by visible light to the acridine orange-DNA complex. Biophys. J. 1:389-400.
219. Bellin, J.S., and C.A. Yankus. 1966. Effects of photodynamic degradation on the viscosity of deoxyribonucleic acid. Biochem. Biophys. Acta 112:363-371.
220. Piette, J., C.M. Calberg-Bacq, and A. Van de Vorst. 1979. Production of breaks in single- and double-stranded forms of bacteriophage FC174 DNA by proflavine and light treatment. Photochem. Photobiol. 30:369-378.
221. Doudney, C.O. 1967. Photoreversal of acriflavine inhibition of DNA replica tion in UV-exposed bacteria. Photochem. Photobiol. 6:651-656.
222. Friefelder, D. 1966. Acridine orange- and methylene blue-sensitized in duction of Escherichia coli lysogenic for phage lambda. Virology 30:567-568.
223. Cadet, J. 1986. Photosensitized reactions of nucleic acids. Biochimie 68:813-834.
224. Calberg-Bacq, C.M., F. Siquet-Descans, and J. Piete. 1977. Photodynam ic effects of proflavine on bacteriophage FC174 and its isolated DNA. Pho tochem. Photobiol.26: 573-579.
225. Martens, B. and M. Clayton. 1977. Strand breakage in solutions of DNA and ethidium bromide exposed to visible light. Nucl. Acids Res. 5:1393-1407.
226. Piette, J. and P.D. Moore. 1982. DNA synthesis on FX174 template dam aged by proflavine and light treatment. Photochem. Photobiol. 35:705-708.
227. Simon, M.I., and H. Van Vunakis. 1962. The photodynamic reaction of me thylene blue with deoxyribonucleic acid. J. Mol. Biol. 4:488-499.
228. Simon, M.I., and H. Van Vunakis. 1962. The photodynamic reaction of me thylene blue with deoxyribonucleic acid. J. Mol. Biol. 4:488-499.
229. Jacob, H.E. 1971. In vivo production of DNA single-stranded breaks by photodynamic action. Photochem. Photobiol. 14:743-745.
230. Regan, J.D., and Setlow, R.B.1977. The effect of proflavine plus visible light on the DNA of human cells. Photochem. Photobiol. 25:345-346.
231. Crick, F.H.C. 1961. General nature of the genetic code for proteins. Nature 192:1227-1232.
232. Ritchie, D.A. 1964. Mutagenesis with light and proflavin in phage T4. Ge net. Res. Camb. 5:168-169.
233. Imray, P., and D.G. MacPhee. 1973. Induction of frameshifts and base-pair substitutions by acridine orange plus visible light in bacteria. Mutation Res., 20:433-435.
234. Webb, R.B., and H.E. Kubitschek. 1963. Mutagenic and antimutagenic ef fects of acridine orange in Escherichia coli. Biochem. Biophys. Res. Comm. 13(2):90-94.
235. Gutter, B., W.T. Speck, and H.S. Rosenkranz. 1977. A study of the photoin duced mutagenicity of methylene blue. Mutation Res. 44:177-182.
236. Zampieri, A., and J. Greenberg. 1965. Mutagenesis by acridine orange and proflavine in Escherichia coli strain S. Mutation Res. 2:552-556.
237. Nakai, S., and T. Saeki. 1964. Induction of mutation by photodynamic ac tion in Escherichia coli. Genet. Res. Camb.5:158-161.
238. Ito, T., and K. Kobayashi. 1976. Photochem. Photobiol.25:399-401.
239. Meier, H. Photochemistry of dyes. In The Chemistry of Synthetic Dyes, vol. IV, pp.389-515. Venkatarman, K., ed. Academic Press, New York.
240. Christopher Foote. 1974. Photosensitized oxidation and singlet oxygen. In Free Radicals in Biology, Volume II. W. A. Caughey, ed. Academic Press, New York.
241. Adelman, A.H., and G. Oster. 1956. Long-lived states in photochemical reactions. II. Photoreduction of fluorescein and its halogenated derivatives. J. Amer. Chem. Soc. 78:3977-3980.
242. Kellman, A. 1974. Primary photochemical processes of cationinc acridine orange in aqueous solution studied by flash photolysis. Photochem. Photo biol. 20:103-108.
243. Kasche, V., and L. Lindqvist. 1965. Transient species in the photochemis try of eosin. Photochem. Photobiol. 4:923-933.
244. Nilsson, R., P.B. Merkel, and D.R. Kearns. 1972. Unambiguous evidence for the participation of singlet oxygen in photodynamic oxidation of amino acids. Photochem. Photobiol. 16:117-124.
245. Kraljic, I., and L.Lindqvist. 1974. Laser photolysis study of triplet eosin and thionine reactions in photosensitized oxidations. Photochem. Photobiol. 20:351-355.
246. Hasty, N., P.B. Merkel, P. Radlick, and D.R. Kearns. 1972. Role of azide in singlet oxygen reactions: reaction of azide with singlet oxygen. Tetrahe dron Letters 1:49-52.
247. Reszka, K., P. Kolodzie, P.G. Tsoungas, and J.W. Lown. 1988. Photosen sitziation by antitumor agents-6. Production of superoxide radical and hy drogen peroxide during illumination of diaminoanthrcenediones in the pres ence of NADH in aqueous solutions: an EPR study. Photochem. Photo biol.47(5):625-633.
248. Fahey, R.C., W.C. Brown, W.B. Adams, and M.B. Worsham. Occurrence of glutathione in bacteria. J. Bacteriol. 133:1126-1129.
249. Ito, T. 1978. Cellular and subcellular mechanisms of photodynamic action: the singlet oxygen hypothesis as a driving force in recent reasearch. Pho tochem. Photobiol. 28:493-508.
250. Sharpatyi, V.A., and I. Kraljic. 1978. Detection of singlet oxygen in radioly sis of aerated aqueous solutions. Photochem. Photobiol. 28:587-590.
251. Wakayama, Y., Takagi, M., and Yano, K. 1980. Photosensitized inactivation of E. coli cells in toluidine blue-light system. Photochem. Photobiol. 32:601-605.
252. Pottier, R., R. Bonneau, and J. Joussot-duBien. 1975. pH dependence of singlet oxygen production in aqueous solutions using toluidine blue as a photosensitizer.Photochem. Photobiol. 22:59-61.
253. Kawanishi, S., S. Inoue, and S. Sano. 1986. Photodynamic guanine modifi cation by hematoporphorin is specific for single-stranded DNA with singlet oxygen as a mediator. J. Biol. Chem. 261(13):6090-6095.
254. Houba-Herin, N., C.M. Calberg-Bacq, J. Piette, and A. Van de Vorst. 1982. Mechanisms for dye-mediated photodynamic action: singlet oxygen production, deoxyguanosine oxidation and phage inactivating efficiencies. Photochem. Photobiol. 36:297-306.
255. Luttrull, D.K., O. Valdesag, S.M. Linden, J. Paczkowska, and D.C. Neck ers. 1988. Rose bengal aggregation in rationally synthesized dimeric sys tems. Photochem. Photobiol. 47(4):551-557.
256. Paczkowska, B., J. Paczkowska, and D.C. Neckers. 1985. Properties of Rose bengal:(X) new derivatives of polymer-rose b. Photochem. Photo biol. 42(5):603-604.
257. Foote, C.S. 1979. Detection of singlet oxygen in complex systems: a cri tique. In Biochemical and Clinical Aspects of Oxygen, W.S. Caughey, ed. Academic Press: New York.
258. Kobayashi, K. 1978. Effect of sodium azide on photodynamic induction of genetic changes in yeast. Photochem. Photobiol. 28:535-538.
259. Kraljic, I. 1986. Detection of singlet oxygen and its role in dye-sensitized photoxidation in aqueous and micellar solutions. Biochimie 68(6):807-812.
260. Kobayashi, K., and T. Ito. 1976. Further in vivo studies on the participation of singlet oxygen in the photodynamic inactivation and induction of genetic changes in saccharomyces cerevisiae. Photochem. Photobiol. 23:21-28.
261. Saito, I., K. Inoue, and T. Matsuura. 1975. Occurrence of the singlet oxy gen mechanism in photodynamic oxidations of guanosine. Photochem. Photobiol. 21:27-30.
262. Lee, P.C.C., and M.A.J. Rodgers. 1987. Laser flash photokinetic studies of rose bengal sensitized photodynamic interactions of nucleotides and DNA. Photochem. Photobiol. 45(1):79-86.
263. Nilsson, R., P.B. Merkel, and D.R. Kearns. 1972. Unambiguous evidence for the participation of singlet oxygen in photodynamic oxidation of amino acids. Photochem. Photobiol. 16:117-124.
264. Wakayama, Y., Takagi, M., and Yano, K. 1980. Photosensitized inactivation of E. coli cells in toluidine blue-light system. Photochem. Photobiol. 32:601-605.
265. Ito, T. 1978. Cellular and subcellular mechanisms of photodynamic action: the singlet oxygen hypothesis as a driving force in recent reasearch. Pho tochem. Photobiol. 28:493-508.
266. OhUigin, C., D.J. McConnell, J.M. Kelly, and W.J.M.vander Putten. 1987. Methylene Blue photosensitized strand cleavage of DNA: effects of dye binding and oxygen. Nucl. Acids Res. 15(18):7411-7427.
267. Reszke, K., and C.F.Chignell. 1983. Spectroscopic studies of cutaneous photosensitizing agents-IV. The photolysis of benoxaprofen, an anti-in flammatory drug with phototoxic properties. Photochem. Photobiol. 38:281-291
268. Kasche, V. 1967. Radical intermediates in the fluorescein- and eosin-pho tosensitized autoxidation of L-tyrosine. Photochem. Photobiol. 6:643-650.
269. Kasche, V., and L. Lindqvist. 1964. Reactions between the triplet state of Fluorescein and oxygen. J. Phys. Chem.68:817-823.
270. Srinivasan, V.S., D. Podolski, N.J. Westrick, and D.C. Neckers. 1978.Pho tochemical generation of superoxide by rose bengal and Ru(bpy)3 2+. J. Amer. Chem. Soc.100:6513-6515.
271. C.W.Jefford and A.F.Boschung. 1976. Dye-sensitized photooxidation of biadmantylidene. Tetrahedron Letters 51:4771-4774.
272. Jahnke, L.S., and A.W. Frenkel. 1978. Photoxidation of epinephrine sensi tized by methylene blue-evidence for the involvement of singlet oxygen and of superoxide. Photochem. Photobiol. 28:517-523.
273. Koizumi, M., and Y. Usui. 1972. Fundamental Aspects of the Oxidative and Reductive Photobleaching of Xanthene and Thiazine dyes. Mol. Photo chem. 4(1)57-92.
274. Peters, G. and M.A.J. Rodgers. 1981. Single-electron transfer from NADH analogues to singlet oxygen. Biochim. Biophys. Acta 637:43-52.
275. Motten, A.G., G.R. Buettner, and C.F. Chignell. 1985. Spectroscopic stud ies of cutaneous photosensitizing agents-VIII. A spin-trapping study of light induced free radicals from chlorpromazine and promazine. Photo chem. Photobiol. 42(1):9-15.
276. Cannistraro, S., and A. Van de Vorst. 1977. ESR and optical absorption ev idence for free radical involvement in the photosensitizing action of furo coumarin derivatives and for their singlet oxygen production. Biochim. Bio phys. Acta 476:166-177.
277. Joshi, P.C., and M.A. Pathak. 1983. Production of singlet oxygen and su peroxide radicals by psoralens and their biological significance. Biochem. Biophys. Res. Commun.112:638-646.
278. Pathak, M.A., and P.C. Joshi. 1984. Production of active oxygen species by psoralens and ultraviolet radiation. Biochim. Biophys. Acta 798:115-126.
279. Saito, I., K. Inoue, and T. Matsuura. 1975. Occurrence of the singlet oxy gen mechanism in photodynamic oxidations of guanosine. Photochem. Photobiol. 21:27-30.
280. Ananthaswamy, H.N., and A. Eisenstark. 1976. Near-UV-induced breaks in phage DNA: sensitization by hydrogen peroxide (a tryptophan photopro duct). Photochem. Photobiol. 24:439-442.
281. Beuttner, G.R., T.P. Doherty, and T.D. Bannister. 1984. Hydrogen peroxide and hydroxyl radical formation by methylene blue in the presence of ascor bic acid. Radiat. Environ. Biophys 23:235-243.
282. Buettner, G.R., and M.J. Need. 1985. Hydrogen peroxide and hydroxyl free radical production by heatoporphorin derivative, ascorbate, and light. Can cer Letters 25:297-304.
283. Koizumi, M., and Y. Usui. 1972. Fundamental Aspects of the Oxidative and Reductive Photobleaching of Xanthene and Thiazine dyes. Mol. Photo chem. 4(1)57-92.
284. Sammertano, L.J., and R.W. Tuveson. 1984. The effects of exogenous ca talase on broad-spectrum near-UV (300-400nm) treated Escherichia coli cells. Photochem. Photobiol. 40(5):607-612.
285. Piette, J., C.-M. Calberg-Bacq, and A. Van de Vorst. 1981. Alteration of guanine residues during proflavine mediated photosensitizing of DNA. Photochem. Photobiol.33:325-333.
286. Black, H.S., W. Lenger, J. Gergius, and V. McCann. 1984. Studies on the photoprotective mechanism of butylated hydroxytoluene. Photochem. Pho tobiol. 40(1):69-75.
287. Buettner, G.R. 1985. Hematoporphorin derivative and light produces the vitamin E radical. In Primary photo-processes in biology and medicine, R.V. Bensasson, G. Jori, E.J. Land, and T.G. Truscott, editors. Plenum Publishing Company.
288. Rizzuto, F., and J.D. Spikes. 1977. The eosin-sensitized photoxidation of substituted phenylalanines and tyrosines. Photochem. Photobiol. 25:465-476.
289. Beuttner, G.R., T.P. Doherty, and T.D. Bannister. 1984. Hydrogen peroxide and hydroxyl radical formation by methylene blue in the presence of ascor bic acid. Radiat. Environ. Biophys. 23:235-243.
290. Buettner, G.R., and M.J. Need. 1985. Hydrogen peroxide and hydroxyl free radical production by heatoporphorin derivative, ascorbate, and light. Can cer Letters 25:297-304.
291. Merville, M.P., J. Decuyper, M. Lopez, J. Piette, and A. Vandervorst. 1984. Phototoxic potentialities of tartrazine: screening tests. Photochem. Photo biol. 40(2):221-226.
292. Van Steveninck, J., K. Tijssen, J.P. Boegheimer, J. Vanderzen, and T.M. Dubbelman. 1986. Photodynamic generation of hydroxyl radicals by hema toporphorin derivative and light. Photochem. Photobiol. 44(6):711-716.
293. Sanford, KK., R. Parshad, and Raymond Gantt. 1986. Genetic Influences on chromosomal radiosensitivity, DNA repair and cancer susceptibility. ATCC Quarterly Newsletter 6(4):1,2,6.
294. Ueda, K., J. Morita, T. Komano, and S. Kobayashi. 1985. Site-specific DNA damage caused by lipid peroxidation products. Biochim. Biophys. Acta 824:341-8.
295. Fujimoto, K., W.E.Neff, and E.N.Frankel. 1984. The reaction of DNA with lipid oxidation products, metals, and reducing agents. Biochim. Biophys. Acta 795:100-7.
296. Santus, R., and J.-P. Reyftmann. 1986. Photosensitization of membrane components. Biochimie 68(6):843-848.
297. Spikes, J.D., and H.M. Swartz. 1978. International conference on singlet oxygen and related species in chemistry and biology review and general discussion. Photochem.Photobiol. 28:921-933.
298. Bezman, S.A., P.A. Burtis, T.P.J. Izod, and M.A. Thayer. 1978. Photody namic inactivation of E. coli by rose bengal immobilized on polystryrene beads. Photochem. Photobiol. 28:325-330.
299. Wakayama, Y., Takagi, M., and Yano, K. 1980. Photosensitized inactivation of E. coli cells in toluidine blue-light system. Photochem. Photobiol. 32:601-605.
300. Nakai, S., and T. Saeki. 1964. Induction of mutation by photodynamic ac tion in Escherichia coli. Genet. Res. Camb.5:158-161.
301. Spikes, J.D., and R. Straight. 1981. The sensitized photoxidation of biomo lecules. In Oxygen and Oxy-Radicals in Chemistry and Biology, M.A.J. Rodgers and E.L. Powers (eds.). Academic Press, New York.
302. Fridovich, I. 1985. Superoxide dismutases: regularities and irregularities. Harvey Lectures 79:51-75.